
(Samajweekly) Thalassemia is a group of inherited blood disorders characterized by defective production of hemoglobin which is the main oxygen-carrying protein in red blood cells.The above cited condition leads to a chronic anemia which could be defined as reduced oxygen delivery supply to tissues and various systemic complications. It is becoming a major health complications in the world.
CAUSES OF THALASSAEMIA
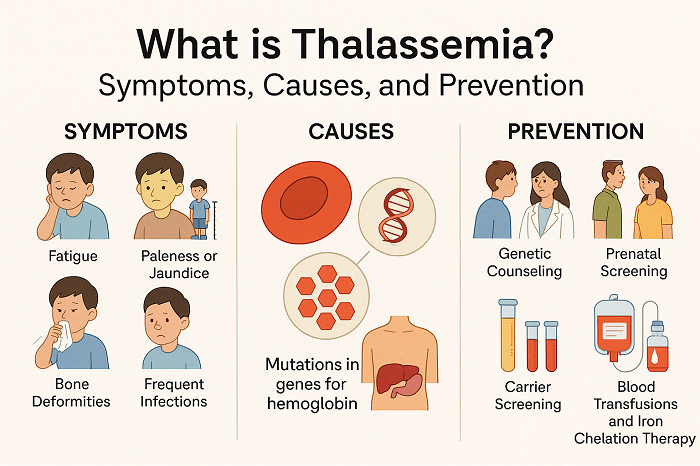
Thalassaemia is caused by faulty genes that could play a vital role in affecting the production of haemoglobin.A child can only be born with thalassaemia if they inherit these faulty genes from both parents.For example, if both parents have the faulty gene that causes beta thalassaemia major, there’s a 1 in 4 chance of each child they have being born with the condition.The parents of a child with thalassaemia are usually carriers. This means they only have 1 of the faulty genes.
CLASSIFICATION OF THALASSEMIA
1. Thalassemia Minor (Trait): This is a carrier state usually asymptomatic with mild microcytic anemia.
2. Thalassemia Intermedia: This is moderate stage anemia requiring occasional transfusions with variable clinical manifestations.
3. Thalassemia Major (Cooley’s Anemia): Severe anemia presenting in early childhood requiring lifelong blood transfusions and medical management.
CLINICAL MANIFESTATIONS
Patients with severe thalassemia exhibit the following traits and symptoms:
1.Pallor, fatigue and weakness due to anemia.
2.Growth retardation and delayed puberty.
3.Bone deformities (especially facial bones) from marrow expansion.
4.Iron overload complications affecting heart, liver and endocrine glands (from repeated transfusions).
DIAGNOSTIC APPROACHES
Diagnosis of the thallasemia involves the following blood tests and screening:
1.Complete blood count (CBC): Shows microcytic hypochromic anemia.
2.Hemoglobin electrophoresis or HPLC: Detects abnormal hemoglobin fractions.
3.DNA analysis: Identifies specific gene mutations.
4.Prenatal screening: Performed in high-risk couples for early detection.
MANAGEMENT AND TREATMENT
1.Regular Blood Transfusions: Regular blood transfusions could corrects anemia but this treatment leads to iron overload.
2.Iron Chelation Therapy: Medications such as deferoxamine, deferasirox or deferiprone could be used to remove excess iron.
3.Bone Marrow or Stem Cell Transplantation: The only curative option especially in young patients with matched donors.
4.Supportive Care: Folic acid supplementation, vaccination and monitoring for infections.
5. Gene Therapy (Emerging): Experimental therapies targeting correction of defective genes hold promise for future cure.
FACTORS EXACERBATING THE CONDITION
1.Lack of access to regular transfusion facilities.
2.Poor compliance with iron chelation therapy.
3.Co-inheritance of other hemoglobinopathies (e.g., sickle cell disease).
4.Infections and malnutrition aggravating anemia.
PREVENTION AND CONTROL
1.Genetic Counseling: Genetic Counseling is must and essential for at-risk couples.
2.Population Screening Programs: Screening a large percentage of population is required especially in high-prevalence areas could help in early detection.
3.Prenatal Diagnosis: Prenatal Diagnosis helps in prevention of transmission of deadly disease to future generations.
4.Awareness Campaigns: Awareness drives could help in reducing the stigma and it could also promote early diagnosis.
Thalassemia is not just a hematological disorder but it is defined as a lifelong genetic challenge that could severely impact the patients, their families and the healthcare systems across the globe.The huge advances and development in the field of the medical management, the bone marrow transplantation and the gene therapy are bringing new hope for the patients of thallasemia. However, prevention through awareness, genetic counseling and early screening remains the most effective strategy in reducing the global burden of this disease.
SURINDERPAL SINGH
FACULTY IN SCIENCE
SRI AMRITSAR SAHIB PUNJAB.

 – A Life of Defiance and Discovery From Rebellion to Buddhist Awakening")




